
Portada de la película dirigida y protagonizada por Stanley Tucci en el año 2000.
Si he decidido poner este título a esta sexta entrega de “12 síndromes sin piedad” no es por capricho, bien saben que las películas pretenden siempre servir de nexo entre lo que ellas nos narran y lo que yo, más humildemente, pasaré a exponerle en breves líneas. De esta forma, si recuerdan un poco, el título de este breve es casi idéntico al de la tercera obra cinematográfica de Stanley Tucci: “El secreto de Joe Gould”. La película es un drama centrado en la época neoyorquina en que artistas, escritores y demás personajes relevantes del mundo de las letras y las artes escénicas se congregan en el Greenwich Village para tratar diferentes temas de actualidad. Aquí se mueve como pez en el agua el escritor Joseph Mitchell, encarnado por el propio Tucci, y este será el lugar conocerá al andrajoso Gould, un “sabio” que se ha doctorado en las calles de la mencionada ciudad estadounidense. La obra de Gould, que lleva por nombre “Historia oral de nuestro tiempo”, en la que recoge y transcribe cientos de conversaciones y ensayos sobre todo lo que ha visto u oído en sus encuentros con otros doctos prosistas y literatos, acaba convirtiéndose en una obra de referencia gracias al apoyo de Mitchell, quien escribe en The New Yorker sobre tan excelente libro y extravagante autor, que finalmente acaba ganándose el apelativo de celebridad.
“Historia oral de la ciencia de nuestro tiempo” podría llamarse la biografía conjunta de Harris, Autio y Sanfilippo. Imagino que los nombres no les sonarán, ya que no han militado nunca en las filas del Real Madrid ni han batido ningún récord mundial. Estos señores son los primeros descriptores de una enfermedad denominada mucopolisacaridosis tipo III, también conocida como síndrome de Sanfilippo. Se conoce también de esta manera debido a que en 1962, el propio Sanfilippo diagnosticó a ocho niños con retraso mental y mucopolisacaduria de heparán sulfato, y la denominó de la manera antes mencionada debido a que éstos excretaban este peculiar azúcar en grandes cantidades por la orina, tal como describió un año antes su colega Harris. ¿Ven cómo las piezas van encajando? Ahora es momento de ir desgranando qué enmascara este síndrome.
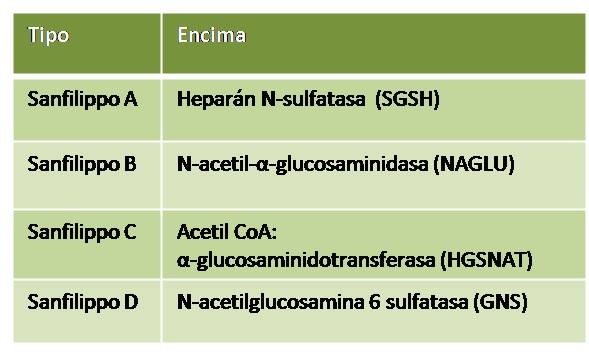
Esquema que recoge los diferentes subtipos de Sanfilippo descritos hasta la fecha y qué enzima se implicada se encuentra defectuosa en cada uno de ellos.
Bajo el síndrome de Sanfilippo se conoce a uno de los múltiples trastornos metabólicos de carácter hereditario donde el organismo no es capaz de descomponer correctamente las cadenas de azúcares denominados glucosaminoglicanos, biomolécula de función estructural que se halla en el tejido epitelial u óseo entre otros. En concreto, ocurre por la deficiencia o carencia en las enzimas necesarias para descomponer las cadenas de heparán sulfato, de tal manera que, dependiendo de si la hidrolasa lisosomal implicada en las diferentes etapas de degradación está afectada o no, tendremos un subtipo u otro de síndrome de Sanfilippo, siendo la forma más grave a la par que de mayor incidencia la denominada como subtipo A.

Familia con sus dos hijos afectados por síndrome de Sanfilippo. Observen los rasgos faciales toscos a los que hacían referencia Autio.
Los síntomas que caracterizan a los enfermos de mucopolisacaridosis tipo III abarcan un espectro muy amplio, pero todos ellos tienen en común que comienzan a manifestarlos a partir del primer año de vida, donde el niño empieza a adquirir unos rasgos faciales “toscos” (aspecto de gárgola, como lo definiera en la década de los 70’s Autio y sus colaboradores) y al que acompaña una disminución en la capacidad de aprendizaje. Conjuntamente a este aspecto reseñable, se suman a esta característica otras tales como dificultad para dormir y para caminar, ya que sus articulaciones se vuelven rígidas y no se extienden por completo. Los casos más severos se complican con ceguera del afectado, la presencia de convulsiones prolongadas en el tiempo y el fallecimiento del individuo a edades tempranas, casi siempre antes de alcanzar la pubertad. De esta forma, el diagnóstico de la enfermedad se realiza por medio de un diagnóstico físico sencillo, ya que estas personas suelen mostrar una inflamación del bazo y el hígado. Además de la elevada cantidad de heparán sulfato en la orina, mencionada líneas arribas, muestran córneas transparentes tras realizarles un examen ocular, técnica que servirá para discriminar entre el síndrome de Sanfilippo (MPS-III) o el síndrome de Hurler (MPS-I H), en el que las córneas muestran una coloración opaca.
Hasta aquí hemos hablado del diagnóstico y a qué se debe en última instancia este fenómeno, pero no hemos explicado la causa subyacente a esta deficiencia enzimática concreta. Obviamente, detrás de la mayoría de los “defectos” se encuentra la Genética y este caso no es especial en este aspecto, más allá de la sintomatología intrínseca al síndrome. El síndrome de Sanfilippo se hereda como un carácter autosómico recesivo, es decir, necesitamos de las dos copias “anormales” del gen para que se desarrolle la citada enfermedad. Si nos centramos más concretamente en el tipo A del síndrome de Sanfilippo, el trastorno está provocado por una mutación en el gen SGSH, el cual se localiza en 17q25.3 y está constituído por ocho exones, los cuales codifican para una N-sulfoglucosamín sulfhhidrolasa, una de las cuatros enzimas anteriormente mencionadas encargadas de degradar el heparán sulfato. Este gen presenta la particularidad añadida de mostrar un patrón de maduración alternativo, de tal manera que se han descrito hasta la fecha tres transcritos diferentes de 3.1, 4.3 y 7.1 kb de tamaño. (Nota aclaratoria: 1 kb = 1.000 pares de bases de ADN o ARN).
Cromosoma 17 humano, donde se indica la zona mutada, desencadenante del síndrome de Sanfilippo.
Para cerrar el breve de hoy, decir que Gloria González Aseguinolaza, ha abierto una puerta a los familiares de muchos de los afectados. Esta investigadora del área de terapia génica y hepatología del Centro de Investigación Médica Aplicada de la Universidad de Navarra apela a la financiación de un ilusionante proyecto de investigación para poder trasladar los resultados obtenidos en ratones por un grupo catalán a humanos. “Se ha visto que la terapia génica funciona muy bien con animales . Hay varios estudios que lo demuestran. El más cercano, en Barcelona, ha dado resultado positivo. Los ratones se recuperan”. La única piedra en el camino son 3 millones de euros. 3 millones de euros separan a 70 familias españolas de poder dar una mejor calidad de vida a sus hijos. Tan convencida está Gloria, que asegura que “si se hace un ensayo clínico ya, el síndrome de Sanfilippo puede tener cura, un tratamiento, en menos de diez años”.

Una vez más, la ciencia en España no necesita de tiempo, sino de inversión. La paciencia es una virtud de los científicos, aunque imagino que los familiares de los afectados discreparán en esto conmigo, ya que a ellos y sus hijos el tiempo se les agota lamentablemente.
Si deseas colaborar económicamente, puedes hacerlo en esta cuenta de Kutxabank: 2095-0028-8091-133-98-728 o puedes ponerte en contacto con la Fundación Stop Sanfilippo a través de su web: www.stopsanfilippo.org.
