
Imagen del neurólogo austríaco Andreas Rett
He decidido titular este último síndrome sin piedad de esta forma por múltiples motivos, como ahora verán. A primera vista, el título puede parecer una reminiscencia del remake del film “Death Takes a Holiday” dirigido en 1998 por Martin Brest y que llevó por nombre en nuestro país el sonoro título de “¿Conoces a Joe Black?” En la citada película, Anthony Hopkins, encarnando al personaje de William Parrish, debe morir a los 65 años. Sin embargo, la muerte, que adopta el porte de Brad Pitt en esta ocasión, le concede un “impasse” ofreciéndole el trato de que no lo llevará al más allá mientras le sirva de instructor en el proceso de conocer cómo es ser un humano. Hasta aquí la descripción o sinopsis del 8º trabajo cinematográfico de Brest, pero como dije, poco tiene que ver ésta con el breve más que en les presento a modo de conclusión de este serial en el que hemos hablado de zombis, metamorfosis humanas e incluso tratado determinados tipos de psicosis.
El motivo por el que he titulado este texto de la manera que ya conocen responde a una cuestión de gran calado en el mundo de la ciencia: la difusión del conocimiento científico, tanto entre la propia comunidad científica como entre la población. No me duelen prendas en reconocer que la comunidad científica a menudo no se molesta en hacer accesible y en divulgar sus descubrimientos y nuevos conocimientos en las distintas disciplinas. Esto que les relato fue justamente lo que le ocurrió al médico y neurólogo austríaco Andreas Rett.
En 1966, Rett describió una serie de síntomas comunes a 22 niñas alemanas y publicó el fruto de sus estudios en una publicación alemana. Lamentablemente, no recibió el reconocimiento esperado y la comunidad médica no tuvo constancia de sus hallazgos a causa de la escasa circulación del artículo. No fue hasta finales de 1983 que el sueco Bengt Hagberg publicó en un revista de gran difusión una revisión del síndrome de Rett que comprendía 35 casos, sacando del ostracismo al de Fürth y dándose a conocer la enfermedad descrita casi dos décadas atrás por aquél. ¡De haber existido en la época nuestro equipo esto no habría pasado! Prosigo, que no es momento ahora de autocomplacencia y autobombo.

Niña llevándose mano a la boca, claro síntoma de la enfermedad de Rett
En efecto, el último síndrome sin piedad va a tratar el síndrome de Rett. Entre las características que definen a este síndrome es común la manifestación en el neonato de un severo retraso mental así como en la adquisición del lenguaje, además de una evidente descoordinación motriz, donde aparecen movimientos estereotipados de las manos tales como retorcerlas o morderlas. Conjuntamente a esta sintomatología se observa una progresiva pérdida de interés por el medio social, desarrollándose autismo, de aparición frecuente en las etapas iniciales de la enfermedad. Como pueden advertir, el síndrome de Rett provoca una grave discapacidad en muchos niveles, lo que hace al enfermo totalmente dependiente de los demás para el resto de su vida.

Cromosoma X mostrando zona mutación del gen MECP2
A pesar de las dificultades que acarrea el estudio de una enfermedad desconocida hasta fechas relativamente recientes y a lo poco común de la misma podemos decir ciertas cosas acerca del síndrome de Rett. El primer hecho a tener en cuenta es que afecta únicamente a las féminas, afectando a 1 de cada 10.000 niñas. ¿Por qué sólo en niñas? Los diferentes estudios desarrollados hasta le fecha han asociado los casos de síndrome de Rett con un defecto en un gen de la proteína 2 de unión a metil-CpG (MeCP2). Este gen se encuentra en el cromosoma X, por lo que al poseer las mujeres dos cromosomas X, aún cuando uno de ellos esté defectuoso, el otro cromosoma X es suficiente para que la niña sobreviva. Por contra, los varones nacidos con este gen defectuoso, al no poseer un segundo cromosoma X que compense este defecto, fallecen de muerte prematura o mediante aborto espontáneo.
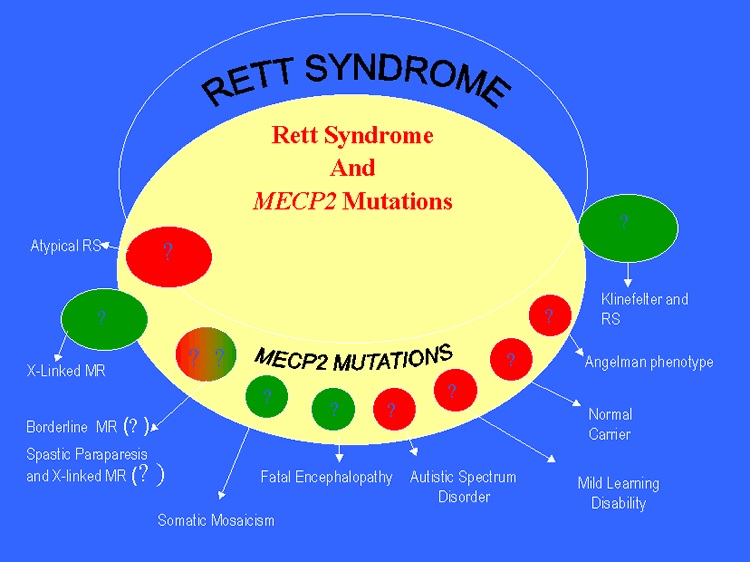
Gráfico con mutaciones MECP2 y sus posibles implicaciones en otras enfermedades
Ya conocemos al culpable de este trastorno, el gen MECP2. Localizado en el brazo largo del cromosoma X (Xq28), dicho gen fue identificado en 1999 por la doctora Amir y sus colaboradores y todos ellos determinaron que codifica para una proteína de unión de ADN (la anteriormente mencionada proteína MeCP2) que tiene como función silenciar a otros genes. Para que todos lo entendamos, podríamos imaginar que MeCP2 es el director de orquesta que con su batuta se encarga de hacer entrar o silenciar a cada uno de los instrumentos que forman parte de la orquesta y por extensión la sinfonía. Lo cierto y verdad es que hasta la fecha se conoce poco más de la proteína MeCP2, tanto cómo funciona como si el síndrome de Rett es el resultado de una larga y desconocida ruta bioquímica de señalización o de qué forma puede estar relacionada con otras enfermedades como el fenotipo Angelman si es que lo está.
Hasta la fecha no existe terapia resolutiva para el síndrome de Rett, tratando el tratamiento tan sólo de contrarrestar el trastorno motor mediante el empleo de fármacos dopaminoagonistas (recordad que la dopamina es el neurotransmisor implicado en la ejecución de movimientos finos y controlados) como la lisurida o la bromocriptina. Muchos médicos e investigadores apuntan a que la terapia definitiva para acabar con esta enfermedad será la terapia génica y se atreven a asegurar que se pondrá a punto en poco tiempo. Sinceramente, pocos somos los que recelamos de creer que esto no sea así, aunque pocos son quienes se atreven a dar una fecha exacta para ello. Desde ésta web, no somos tan osados y tampoco lo vamos a hacer.
De lo que no me cabe duda es de que como decía aquella popular canción de Coz “las chicas tienen algo especial” y de que “son guerreras”, pues el grupo de la doctora Amir ha sido quien ha arrojado más luz sobre esta enfermedad, logrando la tan ansiada caracterización genética de este trastorno.
